WHAT IS ADSL DEFICIENCY?
Adenylosuccinate Lyase (ADSL) deficiency (OMIM: 103050) is a rare, inherited metabolic disorder caused by mutations in the ADSL gene. Adenylosuccinate Lyase is essential for purine biosynthesis and purine nucleotide cycle. Purines are building blocks for nucleic acids (DNA, RNA) and energy-carrying molecules (ATP). Mutations in ADSL gene reduce the activity of the enzyme, leading to an accumulation of two toxic substrates succinylaminoimidazolecarboxamide riboside (SAICAr) and succinyladenosine (S-Ado) in bodily fluids, which results in a wide range of clinical symptoms:
SYMPToMS
Acording to severity, the disease is categorized into three major phenotypes: fatal neonatal type, type I (severe, from wich Minja suffers) and type II (moderate/mild), whereas clinical variability exists even in siblings.
Some of the common symptoms are:
- Decreased fetal activity
- Loss of fetal heart rate variability
- Apneas
- Epilepsy
- Intellectual dissability
- Delayed or absent development
- Autism
- Muscle ataxia
- Hypotonia
- Cortical visual impairement
- Impaired speech
- Hyperactivity
- Microcephaly
INHERITANCE
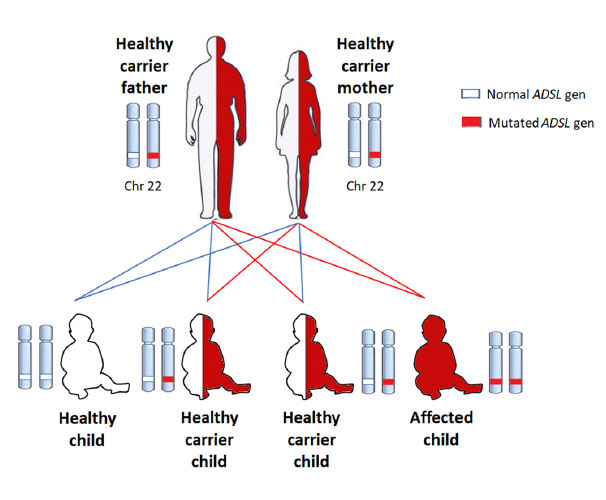
ADSL deficiency is inherited in an autosomal recessive pattern. Parents are healthy carriers with a 25% risk in every pregnancy of having an affected child.
INCIDENcE
The incidence of ADSL deficiency remains unknown. Several hundreds patients are diagnosed worlwide, however, as with other rare diseases, it is estimated that there is a significant number of undiagnosed patients.
DIAGNoSIS
Diagnosis can be acquired via two methods:
Genetic testing – mutation detection in ADSL gene by sequencing with possible characterization of mutant proteins.
Biochemical testing – detection of elevated levels of SAICAr and s-Ado in urine, plasma and/or cerebrospinal fluid.
IS THeRE a THeRAPY?
There is no aproved specific therapy for ADSL deficiency. Patients take anticonvulsants for seizure control, and often require polypharmacy with the use of two or more anticonvulsants. Drug resistence is common.
The lack of effective therapy for this devastating disease made us determined in our mission to connect and colaborate with rare and precious ADSLd research groups, and to initiate and fund critical research which would finnaly lead to helping patients and their families.